AtaGenix Laboratories

AtaGenix Laboratories
Release time: 2025-08-22 View volume: 800
Neuropathic Pain (NP) is a chronic, refractory pain condition caused by peripheral or central nerve injury, often accompanied by emotional disorders such as anxiety and depression. Its mechanisms are complex, involving changes in neuronal excitability, abnormal synaptic plasticity, and multi-level gene expression regulation. Recent studies have shown that epigenetic modifications, such as RNA methylation, play a significant role in neurological disorders. Among these, m⁶Am modification, located near the 5′ cap structure of mRNA, affects mRNA stability and translation efficiency, potentially impacting chronic pain. PCIF1 is known as a key methyltransferase catalyzing m⁶Am modification, while Serpine1 mRNA-binding protein 1 (SERBP1) is an mRNA-binding protein with chaperone functions, critical for mRNA stability, neurogenesis, and synapse formation. However, the roles of PCIF1, SERBP1, and m⁶Am in the development of neuropathic pain remain unclear.
PCIF1 is Significantly Upregulated After Nerve Injury, Specifically in S1HL Excitatory Neurons
To address these questions, the authors conducted systematic studies using various neuropathic pain animal models (SNI, CCI, chemotherapy, or type 2 diabetes models). In the sciatic nerve ligation (SNI) mouse model, m⁶Am modification levels were significantly elevated in the contralateral primary somatosensory cortex hindlimb region (S1HL), accompanied by a marked increase in PCIF1 protein expression. Immunostaining revealed that this upregulation primarily occurred in excitatory neurons in layer 5 of S1HL, while no significant changes in PCIF1 levels were observed in inflammatory pain or chronic stress models. This suggests that peripheral nerve injury specifically induces PCIF1 upregulation in the contralateral S1HL, which may account for the elevated m⁶Am levels. PCIF1 changes are a specific response to nerve injury.
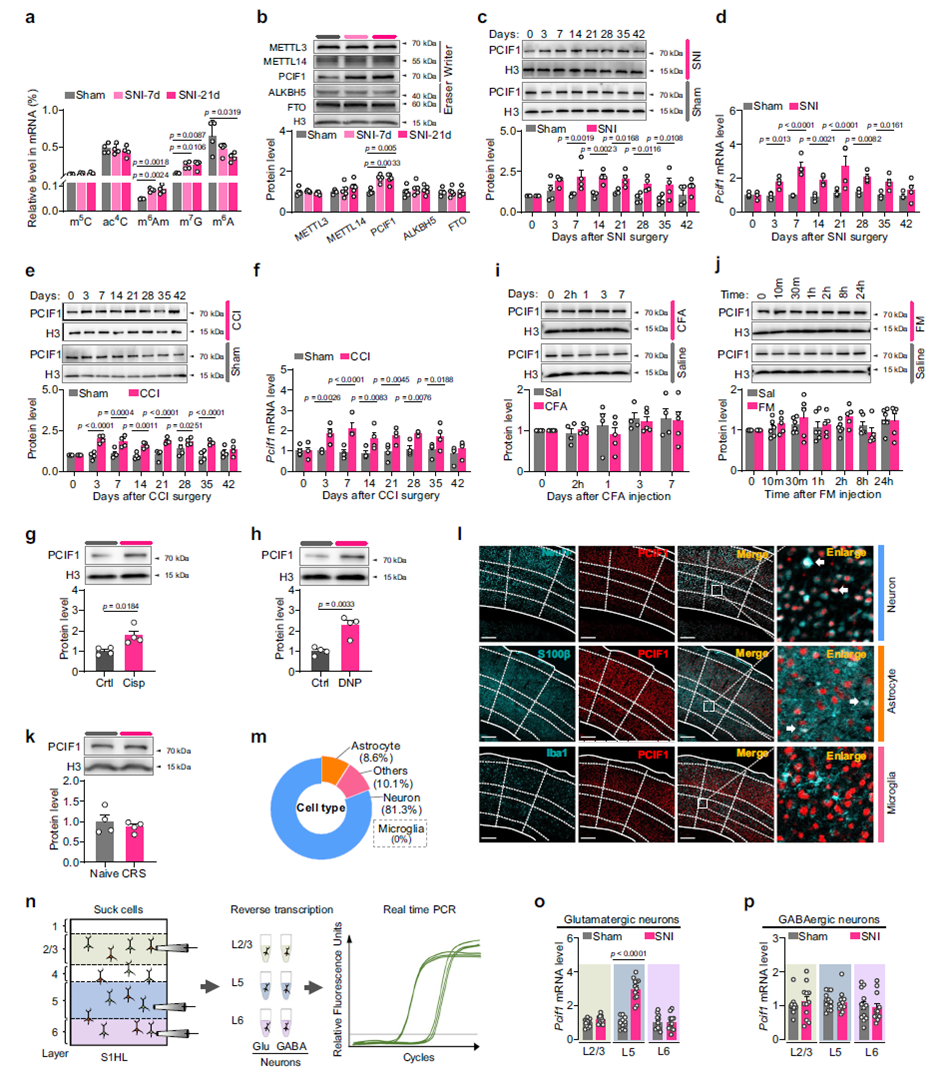
Fig. 1 Neuropathic pain leads to elevated m⁶Am and PCIF1 protein levels in S1HL
Cell-type analysis revealed that PCIF1 upregulation is primarily restricted to glutamatergic (excitatory) neurons in layer 5 of S1HL. Immunofluorescence double-labeling showed that PCIF1 is predominantly expressed in neurons, with no significant presence in microglia. Further experiments using mouse models with specific labeling of excitatory and inhibitory neurons in different layers, combined with single-cell RT-PCR, confirmed that PCIF1 expression increased only in excitatory neurons of the contralateral layer 5 after SNI, with no notable changes in excitatory neurons of other layers or inhibitory neurons across all layers. Laser capture microdissection results further supported this localization.
Inhibiting PCIF1 Alleviates Pain Behaviors and Neuronal Hyperexcitability
To investigate the functional significance of PCIF1 upregulation in S1HL excitatory neurons, the research team specifically knocked down PCIF1 in these neurons. This not only blocked the injury-induced increase in PCIF1 protein but also significantly reduced mechanical and thermal pain sensitivity, indicating that PCIF1 upregulation is critical for the development of neuropathic pain. Further experiments using in vivo calcium imaging and brain slice electrophysiology showed that PCIF1 deficiency eliminated the enhanced neuronal Ca²⁺ signaling and increased firing frequency caused by nerve injury, suggesting a key role in maintaining hyperexcitability of excitatory neurons.
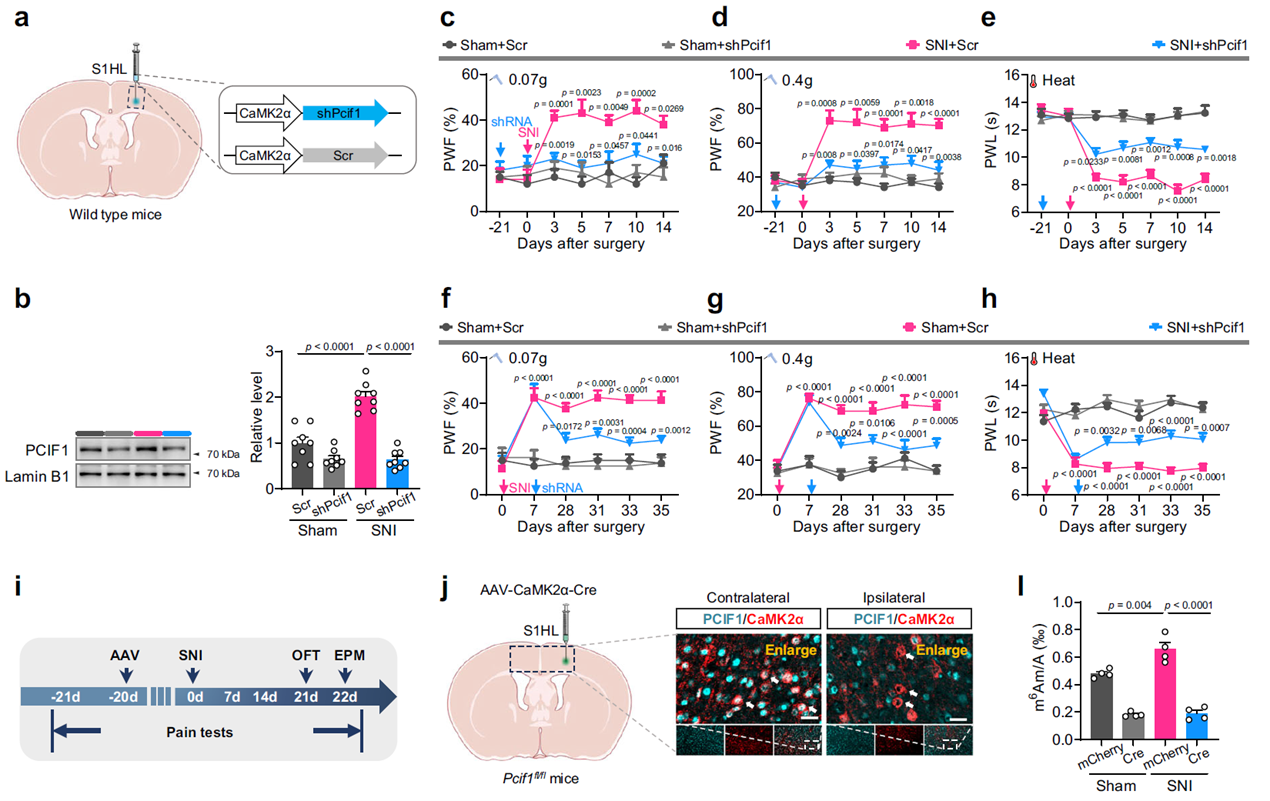
Fig. 2 Increased PCIF1 in S1HL excitatory neurons is essential for the induction and maintenance of neuropathic pain
To determine whether PCIF1 upregulation alone is sufficient to induce a pain state, the research team overexpressed PCIF1 in S1HL excitatory neurons of uninjured mice. These mice exhibited neuropathic pain-like behaviors, including mechanical allodynia, reduced thermal pain thresholds, and anxiety-like behaviors, as well as signs of spontaneous pain in conditioned place preference tests, despite no peripheral nerve injury. This effect was not observed when PCIF1 was overexpressed in the motor cortex, indicating that S1HL is the critical region for this effect.
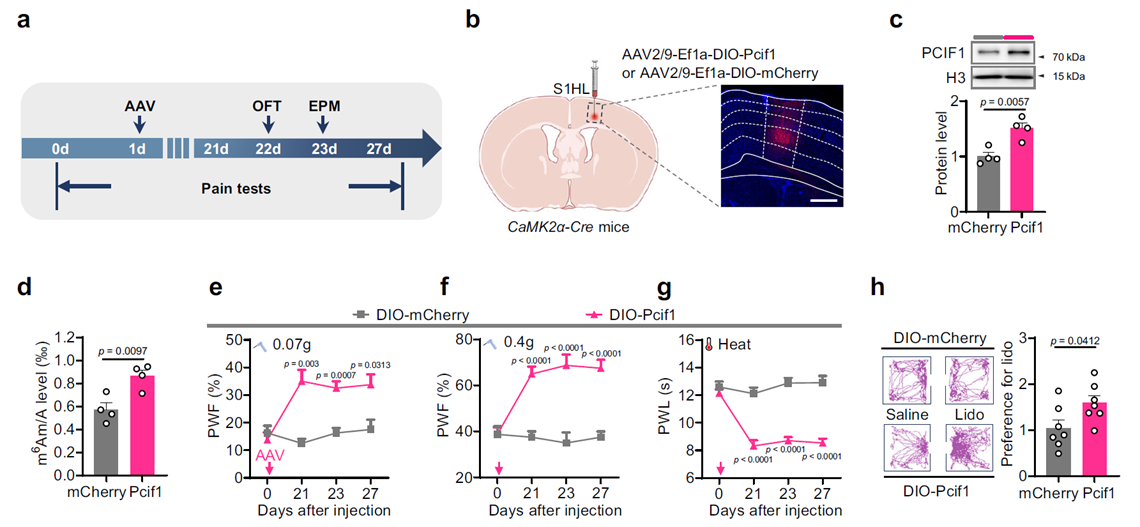
Fig. 3 Upregulation of PCIF1 in S1HL excitatory neurons induces neuropathic pain-like behaviors
PCIF1 Overexpression Directly Induces Pain Phenotypes
At the cellular level, PCIF1 upregulation significantly enhanced the excitability of S1HL excitatory neurons. In uninjured mice injected with AAV-DIO-Pcif1 and AAV-DIO-GCaMP6s in S1HL, facial pain stimulation triggered stronger Ca²⁺ signaling responses. Brain slice recordings also showed that these neurons exhibited significantly higher firing frequencies under depolarizing stimuli compared to controls. These findings suggest that PCIF1 upregulation directly promotes depolarizing responses and firing activity in excitatory neurons, potentially driving abnormal excitation in the pain circuitry.
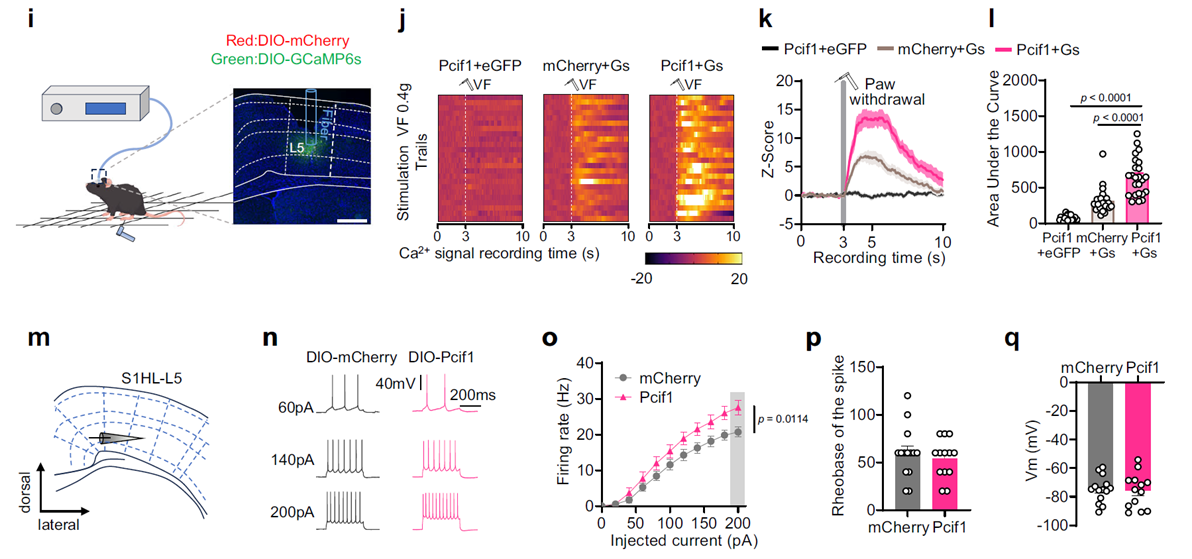
Fig. 4 PCIF1 upregulation directly drives abnormal excitation in the pain circuitry
PCIF1 and SERBP1 Form an Enhanced Complex After Nerve Injury
To explore the mechanism of PCIF1 in neurons, the researchers used endogenous PCIF1 co-immunoprecipitation combined with quantitative mass spectrometry, identifying SERBP1 as a primary interacting protein. This interaction was confirmed in cell lines and mouse models, showing that the two proteins form a complex, with binding significantly enhanced after nerve injury, while PCIF1 downregulation blocked this enhancement. These results indicate that PCIF1 and SERBP1 work synergistically in neurons, mediating m⁶Am modification of specific mRNAs to selectively regulate neuronal function.
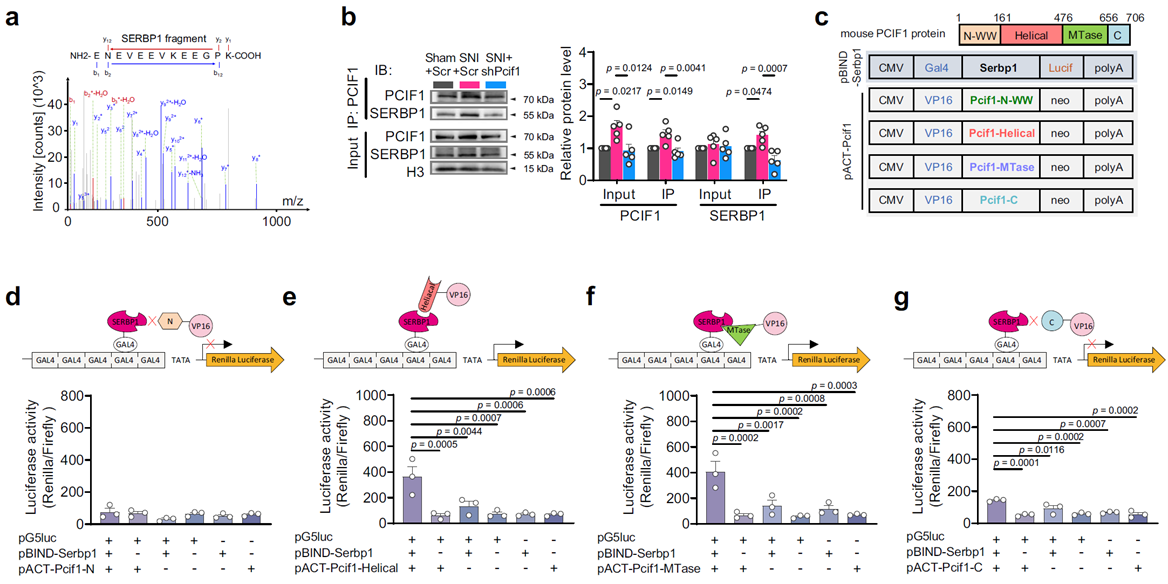
Fig. 5 SERBP1 and PCIF1 interact and jointly catalyze m⁶Am modification on mRNA
In this study, the SERBP1 protein used was custom-produced by AtaGenix using an insect cell expression system, providing critical reagents for validating protein interactions and downstream functions.
Changes in m⁶Am Modification of Target mRNAs and Functional Consequences
To identify downstream targets of the PCIF1–SERBP1 complex, the researchers combined m⁶Am-Exo-Seq and RNA co-immunoprecipitation sequencing, identifying several candidate genes, with Maf1 (RNA polymerase III repressor) mRNA being the most prominent. Maf1 encodes a protein involved in regulating neuronal synaptic structure. Experiments showed that peripheral nerve injury significantly increased m⁶Am modification levels of Maf1 mRNA in S1HL, while knocking out PCIF1 in excitatory neurons completely blocked this change.
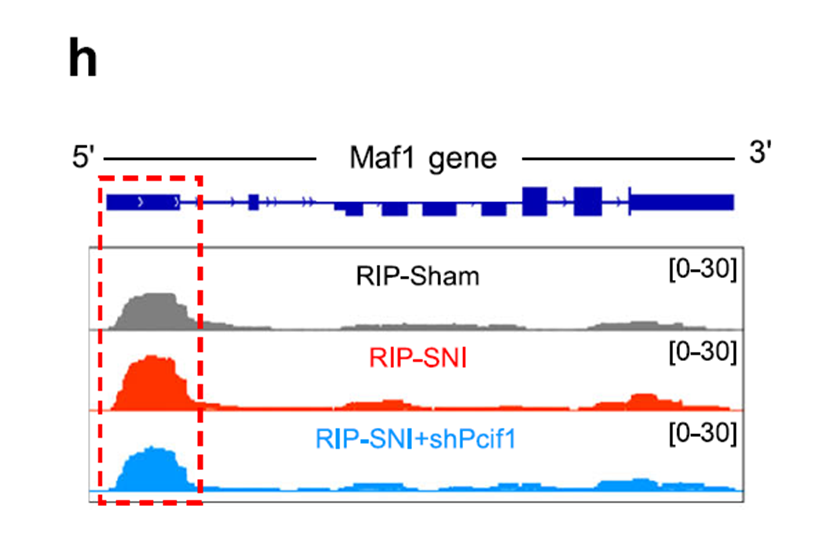
Fig. 6 m⁶Am peak on the Maf1 gene in S1HL
Further experiments revealed that SNI enhanced the binding of PCIF1 to Maf1 mRNA, while PCIF1 deficiency eliminated this enhancement. Additionally, downregulating SERBP1 similarly suppressed the increase in m⁶Am levels of Maf1 mRNA, suggesting SERBP1’s involvement in this modification process. These findings indicate that, under nerve injury conditions, the PCIF1–SERBP1 complex promotes m⁶Am modification of Maf1 mRNA, potentially affecting neuronal function.
PCIF1–SERBP1 Complex Regulates Maf1 mRNA Through m⁶Am Modification
MeRIP-seq and validation experiments revealed that Maf1 mRNA is a primary modification target of the PCIF1–SERBP1 complex. In the SNI model, m⁶Am levels of Maf1 mRNA were significantly elevated, while Maf1 protein expression decreased. Knocking down either PCIF1 or SERBP1 blocked these changes, indicating their synergistic role in mediating target mRNA modification.
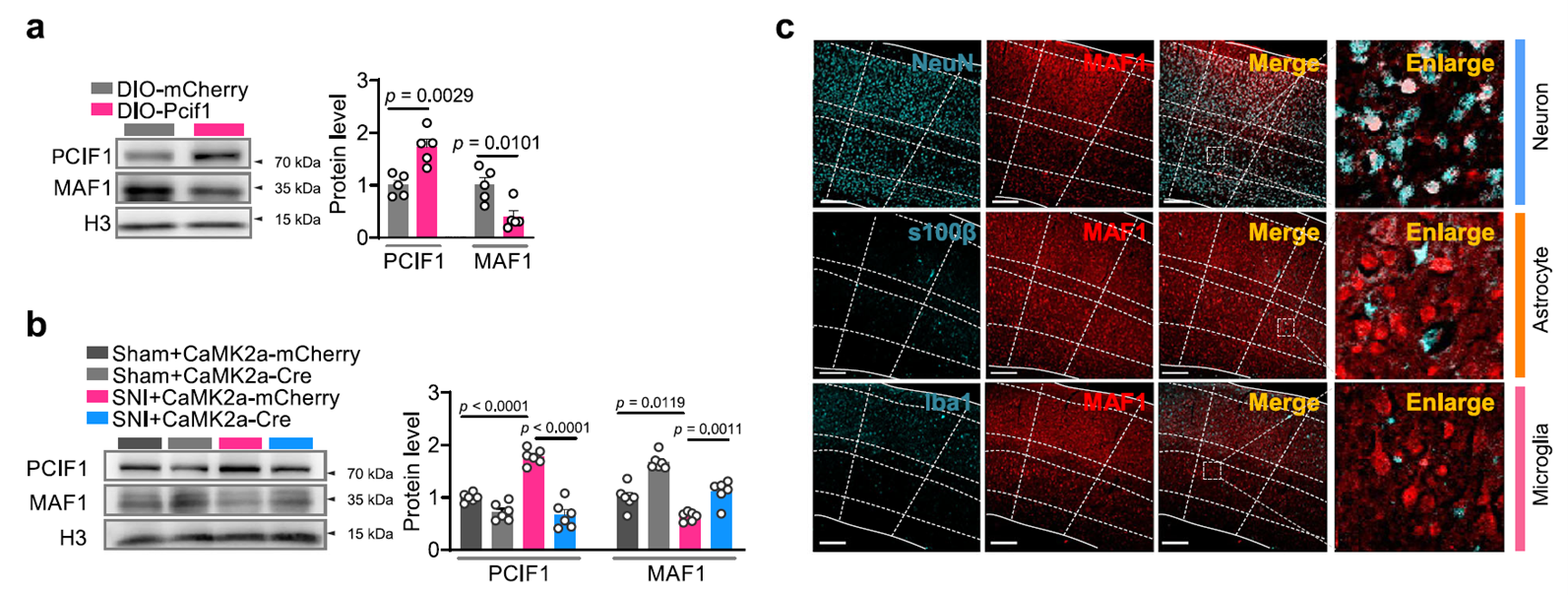
Fig. 7 Increased PCIF1 leads to SNI-induced downregulation of MAF1 in S1HL
MAF1 Functional Validation
Downregulating MAF1 in healthy mice mimicked the pain and neuronal excitability phenotypes induced by PCIF1 overexpression, while restoring MAF1 expression partially reversed the behavioral and electrophysiological changes caused by PCIF1 upregulation. This confirms that MAF1 is a key functional downstream target of the PCIF1–SERBP1–m⁶Am modification pathway.
In uninjured mice, AAV-mediated shRNA knockdown of MAF1 induced mechanical and thermal pain sensitivity, accompanied by enhanced calcium signaling and increased spontaneous firing frequency in S1HL neurons, indicating that MAF1 reduction alone can drive pain-related neuronal changes. In the PCIF1 overexpression model, AAV-mediated restoration of MAF1 expression significantly alleviated pain sensitivity and reduced excessive calcium responses and firing frequency in neurons. These results suggest that MAF1 plays a critical inhibitory role in the PCIF1–SERBP1–m⁶Am pathway, limiting neuronal hyperexcitability and alleviating pain states.
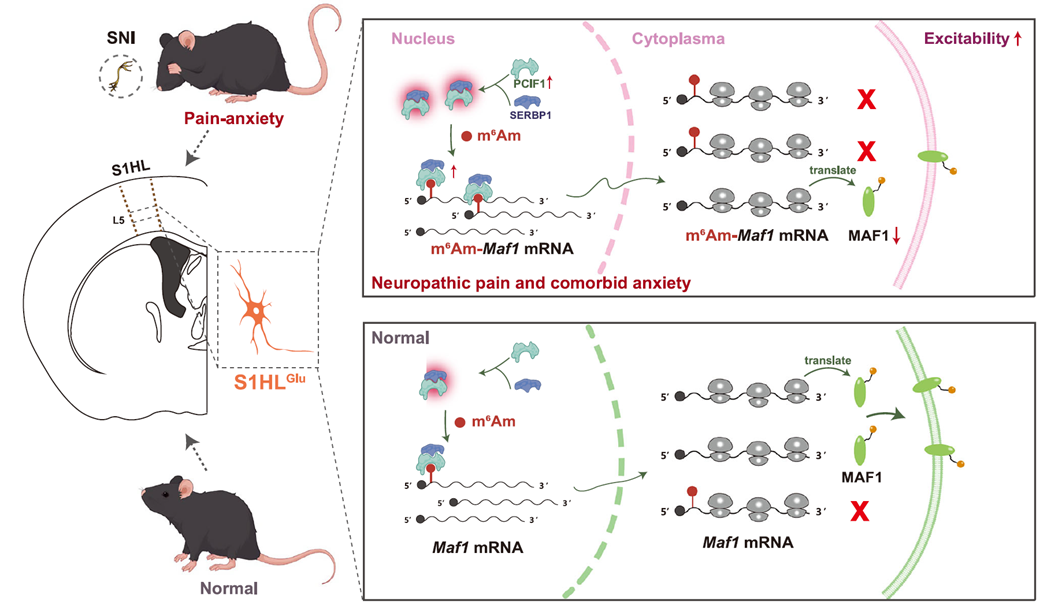
Fig. 8 How the SERBP1-PCIF1 complex in S1HL excitatory neurons contributes to the development and maintenance of neuropathic pain and comorbid anxiety
Overall, this study found that in a mouse model of neuropathic pain with comorbid anxiety, PCIF1 is specifically upregulated in excitatory neurons of layer 5 in the primary somatosensory cortex hindlimb region (S1HL) and interacts with SERBP1 to promote m⁶Am modification at the 5′ end of Maf1 mRNA, thereby suppressing MAF1 protein expression, enhancing neuronal excitability, and inducing pain and anxiety-like behaviors. This suggests that the SERBP1–PCIF1–MAF1 pathway is a key molecular mechanism and potential therapeutic target for neuropathic pain and anxiety.
AtaGenix provided the research team with high-purity, highly bioactive recombinant SERBP1 protein for in vitro m⁶Am catalysis experiments to validate SERBP1’s role in PCIF1-mediated m⁶Am modification. The SERBP1 protein was produced using a baculovirus-insect cell expression system and subjected to rigorous quality control to ensure experimental stability and reproducibility.
To date, over 400 publications have cited AtaGenix’s one-stop protein and antibody development technical services. Looking forward, AtaGenix will continue to fully support scientific research, driving technological innovation and breakthroughs.

Contact Us
+86-27-65523339
info@atagenix.com
Building C, R & D Building, No. 666, Shendun 4th Road, Donghu New Technology Development Zone, Wuhan
